求医网

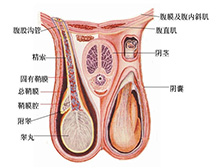
巨睾丸是脆性X染色体即脆性X综合征(fragile X syndrome,FXS)的症状,是一种不完全外显的X染色体连锁显性遗传性疾病。因患者X染色体的短臂Xq27.3带有一脆性断裂点而得名。FXS是一种家族性智力障碍疾病,临床以智力低下、特殊面容、巨睾症、大耳、语言和行为异常为其典型表现。1991年,科学家第一次发现脆性x染色体综合征,它是一种仅次于唐氏综合症的遗传性疾病,是由一不寻常的突变导致的一系列疾病中的一种。
大家都知道,想要尽快恢复健康,除了及时对症治疗并不够,在治疗的同时,建议患者一定要保持愉悦的心情,情绪波动也会影响到此病的治疗,除此之外,生活中的护理也是必不可少的,不仅仅要重视饮食合理,一定要明白该病患者什么可以吃,什么不能食用。最后提醒大家,一旦出现病症,就要及时就医治疗!
来源资料:《解剖学杂志》1994年第6期
巨睾丸是脆性X综合征的表现方式之一。其征X染色体长臂末端脆性位点的发生可能DNA合成代谢过程中的脱氧胸苷-磷酸不足有关,而脆性位点为富有DNA的节段,当脱氧胸苷-磷酸减少时,脱氧胸苷三磷酸减少,这样使在有丝分裂时这一节段不能紧密折叠,甚至出现裂隙或断裂,表现了脆性。以往对脆性X染色体的研究大多侧重于细胞遗传学水平,随着分子生物学研究的不断深入以及在该病中一些特殊遗传规律的发现,在对脆性部位的分类,脆性X染色体的特殊遗传方式及其产生机制方面的研究中取得了许多重要进展,形成了一些新的概念和理论,使人们开始从一个新的水平和角度认识本病。
脆性部位分类:①遗传型脆性部位(h-fra),又称之为罕见型脆性部位;②结构型脆性部位(c-fra),又称为常见型脆性部位。脆性X染色体的遗传特征:过去认为其典型遗传方式呈X连锁隐性遗传,近年发现它的遗传方式非常复杂,具有与一般遗传病完全不同的特殊遗传规律: ①是通过无异常表型的男性携带者又称外显不能(NP)传递的,他们所生的脆性X女儿无异常表现;②在fra(X)家系中智力低下男性患者约占20%,分离率0.4;③表型异常脆性X女性所生的孩子中,分离率为0.5;④表型异常女性的脆性X来自她们的母亲,而非来自父亲;⑤约35%女性携带者出现智力低下;⑥NP男性脆性X母亲一般表型都正常,NP男性子女中出现表型异常的脆性X患者的危险性较低;⑦几乎所有脆性X综合征患儿的母亲都携带脆性X;⑧同胞中的外显程度不一。有关产生机制的几个假说:①可移动因子插入假说;②多聚嘌呤/多聚嘧啶顺序扩增假说;③富含嘧啶DNA顺序重组和扩增假说;④不稳定性前突变假说;⑤常染色体阻抑基因效应假说。
Jacobs等认为男女生殖细胞质的基因可有相同的突变频率,均是向下传递,脆性X染色体患者可能是经过两个阶段基因工程突变而向下传递给父或母,但父或母的表型为正常,在其外周血往往检不出fra(X),当他或她体内生殖细胞再一次发生突变,并传给子或女,即产生了fra(X)阳性患者或女性携带者,Sutherland曾指出,脆性X染色体患者中是否存在异常的基因产物或缺少某些产物?目前尚无依据,总之脆性位点发生的真实机制,尚待更深入的研究。
巨睾丸的发病机制是脆性X染色体病人Xq27.3区带存在脆性位点(FRAXA)是其典型的细胞遗传学特征,1993年脆性X染色体编码基因cDNA被克隆,发现(CGG)n结构中n拷贝数由正常6~52扩增至≥230时,是脆性X染色体患者发病的分子基础,异常扩增的(CGG)n结构位于FMR-I基因翻译区外显子1,至今已知FMRI基因的错义突变和缺失突变携带者也表现出FMRI基因动态突变相同的临床症状,从而显示出脆性X染色体临床症候的患者具有高度的遗传异质性,进而使这些患者及其家庭成员的基因诊断进一步复杂化。
1.细胞遗传学: X染色体长臂27.3带上一个叶酸敏感的部位与脆性X染色体有关,该部位经特殊处理后可显示脆性断裂,因而被称为脆性部位,具有脆性部位的染色体被称为脆性染色体,迄今已有26个染色体的脆性部位被发现,其中仅X染色体X27-X28区域的脆性部位(FRAXA)与遗传性疾病有关,而其他与疾病无关的脆性部位称为普通脆性部位。
X脆性部位产生的机制尚不完全清楚,目前认为与DNA的合成代谢过程有关,已发现在缺乏叶酸或用较大剂量的5-氟尿嘧啶(5-FU)等条件下处理,可致使胸腺核苷合成部分受到遏制,染色体结构就可能在某些特定的部位上产生裂隙或断裂。
2.FMR-Ⅰ基因的结构,转录和翻译: 脆性X染色体的基因称为脆性X智能落后1基因(fragile X mental retardation-Ⅰ,FMR-Ⅰ),定位于Xq27.3区带,在基因组中跨越38kb,由17个外显子和16个内含子组成,FMR-Ⅰ基因的mRNA为4.4kb,编码一个分子量约为69~70kDa,由596个氨基酸组成的脆性智能落后蛋白(FMRP),这是一种RNA结合蛋白,在体内多种组织中都表达。
FMR-Ⅰ基因外显子较小(51~196bp),但内含子较大,平均大小为2.2kb,其中内含子1约为9.9kb,基因中存在多种累及FMR-Ⅰ基因3’端外显子10,12,14,15和17的多种转录拼接形式,其中涉及外显子12和14通常导致整个外显子的丢失,但累及外显子10,15和17的则只丢失这3个外显子5’端的部分序列,这是因为这3个外显子的5’一端序列上分别存在一个拼接保守信号,转录后拼接如发生在这个位置上,则导致这3个外显子5’端的部分序列丢失。
3.FMR-Ⅰ基因的动态突变: FMR-Ⅰ基因的5’端有一个CGG三核苷酸重复区域,在正常个体中CGG结构的重复次数具有多态性,在6~52次,平均为30次,国内人群中以(CGG)28最多见,在FXS患者中,CGG拷贝数一般>200次,多则可达1000次以上,脆性X染色体发生的根本原因是FMR-Ⅰ基因的突变所致,动态突变是指FMR-Ⅰ基因在传递过程中CGG拷贝数不稳定,会发生扩增,这是95%以上的FXS患者发病的分子遗传学基础,动态突变包括3种类型:
(1)FMR-Ⅰ基因的前突变(premutation):FMR-Ⅰ基因(CGG)n结构中n拷贝数扩增至53~230时,携带者虽然表型正常,但在传代过程中易发生进一步的扩增,使后代的CGG重复数大为增加,并有异常表型出现,FMR-Ⅰ基因的这种突变称为前突变,男性或者女性FMR-Ⅰ前突变基因携带者智力水平与正常人并没有区别。
根据统计,男性或者女性携带前突变FMR-Ⅰ基因(CGG)n结构中,n拷贝数没有区别,但随着女性携有的前突变FMR-Ⅰ基因(EGG)n结构中n拷贝数的逐渐增大,在传代过程中扩增至前突变概率也逐渐增加,而38%前突变型FMR-Ⅰ基因经父源性传递至女儿时其(CGG)结构中n拷贝数发生了缩减,但这现象仅见于2%母源性前突变型FMR-Ⅰ基因传递至女儿时,从而提示前突变型FMR-Ⅰ基因在母女传递的过程中其(CGG)n结构中n拷贝数具有扩增的倾向,但父女传递时则存在缩减的趋势。
(2)FMR-Ⅰ基因的全突变(full mutation):FMR-Ⅰ基因由前突变状态(CGG)53~230次扩增至>230次时,100%男性携带者表现为典型的脆性X综合征,53%的女性携带者表现出轻重程度不等的智力低下,此时称为全突变,全突变与智力低下临床表现的出现直接相关,经研究发现,当CGG结构的重复数达230次以上时,FMR-Ⅰ基因5’端的CpG岛开始非正常地甲基化,这种甲基化延伸至启动子区,致使转录不能启动,mRNA不能转录,基因编码的蛋白产物也因之缺乏,导致临床症状产生。
值得指出的是极少数男性全突变型FMR-Ⅰ基因携带者缺乏应有的FRAXA位点脆性现象,其分子遗传学的基础有待于进一步加以研究,就智力低下临床表现而言,几乎100%男性FMR-Ⅰ全突变基因携带者存在智力低下,其中约89%(245/274)为中度智力低下,但仅21%(36/170)女性FMR-Ⅰ全突变携带者表现出中度智力低下,而且高达59%(100/170)女性FMR-Ⅰ全突变型携带者并不出现智力低下。
(3)FMR-Ⅰ基因的回复突变:处于前突变或全突变状态的FMR-Ⅰ基因的CGG结构在传代过程中其拷贝数目会发生一定范围的缩减,称为FMR-Ⅰ基因的回复突变(reverse mutation),根据突变前后FMR-Ⅰ基因所处的状态,回复突变可分为3种类型:全突变型→前突变型;全突变型→全突变或前突变嵌合型;嵌合型或前突变型→正常FMR-Ⅰ基因,这些现象可发生于父源性传递过程,也可发生于母源性传递过程中,虽然较少见,却增加了预测FMR-Ⅰ基因动态突变规律的困难,导致家族内遗传咨询及产前基因诊断进一步复杂化。
4.性别因素对FMR-Ⅰ基因动态突变的影响: 目前认为FMR-Ⅰ基因(CGG)n结构的扩增是一个多途径多步骤递次扩增的过程,脆性X染色体的遗传模式常有特殊的规律,即具表型正常的男性携带者可将脆性部位传递给其女儿,后者一般无智力低下或其他临床症状,但她可将受累染色体传递给后代,使家系中的第三代出现FXS患者,此时第三代男孩具有较明显的智力低下,而女孩往往无明显智力异常,也即由母亲传递给子女所造成的危害较由父亲传递为大,且男孩受累程度较女孩为重,此现象称Sheman现象,此外,还发现男性全突变型FMR-Ⅰ基因携带者精子标本中只存在前突变型FMR-Ⅰ基因,因此,现认为FMR-Ⅰ基因的全突变并未累及男性全突变携带者的生殖细胞,但目前尚缺乏足够的证据表明女性卵细胞也未发生FMR-Ⅰ基因的全突变。
此外,女性FMR-Ⅰ基因动态突变携带者在传代过程中,其(CGG)n结构的扩增和大小尚依后代的性别而发生变化,即传至男性后代时则具有进一步扩增的趋势,但传至女性后代时则扩增程度较小,且尚可见到(CGG)n结构发生缩减的现象,可能女性所携有的另外1条FMR-Ⅰ基因正常的X染色体遏制了女性胚胎早期阶段全突变型FMR-Ⅰ基因(CGG)n结构的进一步扩增,总之,全突变型FMR-Ⅰ基因(CGG)n结构的变化趋势(是扩增,还是缩减)以及变化适度的大小尚受亲代和子代性别的双重影响,因此,在FMR-Ⅰ基因动态突变家族的遗传咨询时必须考虑FMR-Ⅰ基因的这种突变特征。
5.FMR-Ⅰ基因的非动态突变: FMR-Ⅰ基因除了动态突变外,少数病人还会发生碱基置换和缺失等非动态突变,目前已发现1种错义突变和8种缺失型突变,这些突变所导致的临床症状和动态突变相同,但缺乏FRAXA脆性位点这一特征,目前的报道未显示这种突变有热点区。
来源资料:《国际泌尿系统杂志》2007年第5期
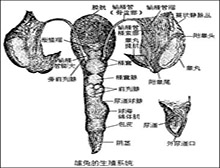
脆性X染色体的临床表现多种多样,性格,心理及精神方面的改变也不完全相同,况且有的患者其临床症状并不十分典型,单靠临床表现很难做出诊断,实验室检查不仅为及早明确诊断提供了可靠的依据,还可以进行携带者的诊断和产前诊断,以及家系调查和群体普查。以细胞学技术检测X脆性位点是一种形态学的检测方法,但准确性和敏感性不是很高,基因检测虽然是诊断FXS的主要手段,但是基因检测不能完全替代染色体检测,因为随着研究的深入,发现脆性X染色体不仅只是与脆性部位FRAXA有关,而且与原来认为是普通脆性部位的FRAXE也相关,近来又发现 FRAXF部位似乎也与脆性X染色体有关,而目前只能检测与FRAXA相关的FMR-Ⅰ基因的突变,所以只进行基因检测容易漏诊。DNA印迹技术可以检测出前突变,嵌合体,全突变以及大片段的缺失,但对较小片段的前突变和缺失则效果较差,PCR则适合于检测重复数小的前突变,但不能检测甲基化。血液检查可以检测出脆性X染色体综合征的患者和携带者。
巨睾丸的鉴别方法如下:
与其他伴有智力低下、特殊面容的遗传性疾病相鉴别,主要依靠实验室检查鉴别诊断。
(1)先天愚型或称Down综合征或21-3体病:本病外貌显示表情呆滞、跟距宽、眼裂小、两眼裂外侧上斜、鼻梁低平、腭弓高、口半张、舌常伸于口外、流涎多。除面容外,手指短、小指内弯、双侧通贯手(即手掌心中的横形纹不分岔而直通连)等。常伴有先天性心脏病、体格及智能发育迟缓。
(2)猫叫综合征:为体细胞内第5号常染色体短臂部分缺失所引起。婴幼儿期哭声似猫叫,特殊面容为头小、脸圆、眼距宽、外眼角下斜、塌鼻梁、耳位低、小下颌,还有通贯手,生长发育迟缓,智力低下严重。
(3)粘多糖病:为粘多糖代谢障碍的一组遗传病,具有丑陋面容,头颅大而呈舟形,前额和两侧颞部突出,颞部发际边缘低,发密粗而直、浓眉、宽眼距、鼻梁低、鼻孔大略上翻、唇厚、张口、舌体大常伸出口外、齿楔形而间距大、颈短、下颌小,角膜混浊从而影响视力,智力迟滞呈渐进性。
(4)海洋性贫血:常染色体遗传病。由于骨髓增生髓腔增宽,面颊骨及颅骨增大,逐渐出现典型的面容,头大、额骨隆起、颧骨高出、鼻梁低平、两服距增宽、面部表情呆滞,生长发育迟缓,智力退化。
(5)呆小病:其特殊外貌为头大、前囟大而囟门闭合较晚、出牙延迟、牙小而稀,因皮下组织有黏液性水肿面部呈水肿样、鼻梁低、眼距宽、眼裂窄、眼睑肿、唇厚、舌大且厚、常伸出口外、流涎、表情呆滞、毛发枯黄而稀疏、皮肤粗糙发干。由于生长发育迟缓故动作笨拙,智力低下。
(6)脑积水:面容有特殊表现,小儿头不能直立,前额向前突出,眼球向下转,上部眼巩膜暴露,呈太阳落山的样子,头皮静脉粗而暴起,前囟扩大而饱满,颅缝裂开,眼球有震颤或斜视,视力减退或消失,头骨与面骨发育不成比例,显出头大而面小,由于脑皮质受压变薄而导致智力低下。
(7)头小畸形:乃指其头围小于同年龄彩虹别的平均值,2岁时头围在42厘米以下。前囟小而近于关闭,面部发育正常而前额及二颞骨向上倾斜,枕部平坦,头颅部显得更小而顶部尖,脑发育受限而导致智力低下。
来源资料:《中华男科学杂志》2002年第6期
脆性X染色体的表现形式之一就是巨睾丸。在巨睾丸诊断确定之后,应设法查明病因。在病史中应询问有无家族史、出生及生长发育情况、有无脑炎、脑膜炎、脑外伤等病史。查体中有无神经系统体征、全身性疾病等。然后选择有关检查,如头颅磁共振(MRI)、CT、血糖、血钙、脑脊液检查等,以进一步查明病因。
巨睾丸的检查方法如下:
1.细胞学检测
(1)脆性X染色体检查:脆性X染色体的检查对于了解脆性部位的表达频率以及脆性部位处染色体的结构非常重要,是确认最初先证者的基本手段。但本方法的检出率较低,一般只有50%左右。由于在X染色体上还存在别的与智力低下无关的脆性部位(如FRQXD等),所以即使检出脆性X位点的存在也不能确诊为FXS患者或携带者。因此,该检查只能作为初筛试验,不能用作确诊的工具。
(2)染色体核型分析:为46,Xq27最常见。
(3)荧光原位杂交技术检查:对于疑为FMR-Ⅰ基因大片段缺失的患者可作FISH检测。以荧光标记的探针,对经过秋水仙素等处理,处于中期分裂象的细胞染色体进行原位杂交,正常染色体有荧光显示,而相应部位有缺失的染色体则无荧光显示。
以细胞学技术检测X脆性位点是一种形态学的检测方法,但准确性和敏感性不是很高。
2.基因检测
(1) DNA印迹技术:可以检测出前突变、嵌合体、全突变以及大片段的缺失,但对较小片段的前突变和缺失则效果较差,PCR则适合于检测重复数小的前突变,但不能检测甲基化。
(2)聚合酶链反应(PCR)技术:选用合适的引物,对患者的FMR-Ⅰ基因片段进行PCR扩增,扩增产物经变性聚丙烯酰胺凝胶电泳分离后直接观察结果,可准确判断CGG的重复数。此法用于发现重复数小的前突变以及观察普通群体中的(CGG)n分布,由此确定正常和前突变之间CGG重复数的分界。该方法简便,适于普查,缺点是CGG重复次数多的全突变顺序中含有大量GC碱基,PCR反应有一定难度。PCR技术无法检出甲基化,故也不能检出嵌合型。约200重复左右的脆性X染色体综合征(约70%).对于更长的需要用southern来检测.
(3)由前突变转化为完全突变只发生母亲向后代传递过程中。根据对脆性部位DNA序列的了解,现已可用RFLP连锁分析、DNA杂交分析、PCR扩增等方法来检出致病基因。
基因检测虽然是诊断FXS的主要手段,但是基因检测不能完全替代染色体检测,因为随着研究的深入,发现脆性X染色体不仅只是与脆性部位FRAXA有关,而且与原来认为是普通脆性部位的FRAXE也相关,近来又发现FRAXF部位似乎也与脆性X染色体有关,而目前只能检测与FRAXA相关的FMR-Ⅰ基因的突变,所以只进行基因检测容易漏诊。
3.蛋白质检测 由于FMRP在正常人几乎每种组织和细胞中均有表达,而在FXS的患者中却不表达或异常表达,因此用抗FMRP单克隆抗体作免疫组化或免疫荧光技术可以检测该蛋白质的存在。早期人们仅对可疑者的血涂片采用此法作检测,近来采用羊水中的胎儿脱落细胞观察是否存在FMRP作为诊断FXS产前诊断的指标。
4.睾丸活组织检查:精细管中精子的发生程度低下,但能正常生殖。
5.常规做B超、心电图、脑电图等检查,可发现大睾丸、脑电图异常波型等。
来源资料:《贵州医药》2002年第4期
巨睾丸为X连锁显性遗传性病,无有效治疗方法。本病征对生命无危害。因为脆性X染色体是一种新认识到的疾病,长远的结果仍未清楚。有脆性X前突变的个体通常不出现临床症状,但是全突变男性伴有精神发育迟滞往往需要中度 到全面帮助。此外,由于交流障碍、行为问题以及社交技能差,他们往往不能独立生活。寿命一般不受到影响。全突变女性长期面对的最主要问题是精神问题,在轻 度认知损害基础上合并的害羞和社会焦虑会明显干扰她们的独立性。因为携带有部分基因变异的女性绝经期开始较早,从而增加了骨质疏松症发生的危险性。
(1)学龄前个性化的治疗计划:
能帮助患儿达到他们的最大潜能。大多患儿可以从医学和特殊教育者团队治疗中获得帮助。这个团队的成员可能包括语音训练师、生理治疗师、职业病治疗师、特殊教育者、心理治疗师以及儿科医生等。接受常规儿科护理,包括免疫接种。另外,眼疾、外表异常、浆液性耳炎、二尖瓣脱垂、癫痫发作和巨睾丸症等问题应在检查过程中评估。眼部疾病包括斜视、近视、脱垂、眼球震颤,最常见的是斜视。如果这种情况发生,折射镜矫正可防止发展成为弱视。涉及矫形外科的问题与相关组织异常有关,包括扁平足、脊柱侧弯、关节松弛等,一般极少需要进行外科治疗。腹股沟疝是相关组织松弛的表现。反复中耳感染需要抗生素治疗,如果这种情况持续,需抽取分泌物和放置引流管,也要进行听力检查。如果有心脏杂音或喀喇声,需要进行超声心动图检查。发现有二尖瓣脱垂需考虑预防性使用抗生素。癫痫,最常见部分发作或全身-阵挛型发作,在约20%发病男性中出现,他们普遍对抗癫痫药反应良好。EEG只能提示是否存在癫痫的可能。大睾丸症可能在青春期前出现,应该让父母认识到这一情况不需要治疗或不会引起任何症状,包括性早熟。一些患儿可以通过改善其行为特征的药物治疗获得帮助,以便能更好的学习。常用的药物是抗抑郁药、刺激性药物(如利他林,用以提高兴奋性)以及抗躁狂药(用以治疗行为和情绪障碍)。
对全突变脆性X染色体的男孩采取的干预措施应针对各种认知、交流和行为损害,可以采用结构化学习环境和行为管理措施治疗多动和刻板行为。视觉文字提示和重复的逐字阅读方法有助于患儿加工新的、程序性信息和视觉-运动协调。计算机学习对促进视觉学习和注意力方面有帮助。此外,这些儿童可从服从简短指令的语言疗法中得益。“塑形”技术(渐进学习法)能很好地帮助他们学习日常生活技能。另外还可从社会技能训练中获益。如果ADHD成为问题,使用精神振奋剂可得到改善。患脆性X染色体女性的教育需要根据认知损害的程度和类型。使用与全突变男性类似的方法对那些有精神发育迟滞表现的女性同样有效。生长发育正常,但有学习困难的女性需要适当的针对非言语性学习障碍的特殊教育服务。个别辅导和精神药物对治疗相关的精神心理障碍有用。她们也可以从社会技能训练中获益。
(2)药物治疗:
临床上使用叶酸治疗本病,鉴于叶酸能通过二氢叶酸还原酶使脱氧胸苷-磷酸增加。给予0.5mg/(kg.d),可使多动、孤独、注意力涣散、不协调运动等有改善,对智力障碍无效。在叶酸治疗好转的过程中,如果应用叶酸代谢阻抑剂时,可使症状恶化,停止应用阻抑剂后再度改善。有研究报道认为叶酸治疗对改善脆性X染色体综合征行为和智力有一定帮助,但国内外研究对叶酸治疗的疗效评价不同,也有研究者认为叶酸治疗的疗效不太理想。 叶酸治疗的根据是发现叶酸能阻止染色体上出现脆性X的改变,但是这种治疗方法是否有效仍存在争论,大部分研究未能证明叶酸对行为和认知有确定的疗效。新近一些作者认为中枢神经兴奋剂疗效较好,但副作用大。其它有用可乐定(clonidine)、心得安者,据称可减轻多动症。
预防巨睾丸的方法:
因为FMRⅠ基因作为脆性X染色体发病基础得到证实,建立了一种特异性更高和成本较低的血液检验(寻找脆性X的DNA试验),可以在全突变和前突变者血液中探测出CGG三核苷酸重复序列的扩张率,这种检查方法可用于产前诊断,专家建议把这一方法用于所有不明原因精神发育迟滞患者。1995年建立了检测FMRP及探查全突变个体血液抗体的方法,这使得筛查脆性X染色体新生儿成为可能。
来源资料:《国内全科医学》2013年第12期


关于我们|招贤纳士|联系我们|用户协议|帮助中心|网站地图|内容合作|友情链接|新浪微博|下载安卓客户端
免责声明:求医网所有建议仅供用户参考。但不可代替专业医师诊断、不可代替医师处方,请谨慎参阅,本站不承担由此引起的相关责任。 Copyright © 2016 QIUYI.CN 京ICP证111012号 公安备案号11011202000697 京ICP备11039101号 互联网药品信息服务资格证书